Sample identification with Kraken
To identify a sample from sequencing reads, we can use the tool “Kraken”. This tool can also be used to identify members in a mixed set of reads, for metagenomics.
-
e.g. reads from one sample → Kraken → 95% Staphylococcus aureus.
-
e.g. mixed reads → Kraken → 50% Staphylococcus aureus, 40% Campylobacter concisus, 10% unclassified.
In this tutorial we will use Kraken to confirm the identify of reads from a bacterial isolate.
Get data
- Log in to your Galaxy instance (for example, Galaxy Australia, usegalaxy.org.au).
Use shared data
If you are using Galaxy Australia, you can import the data from a shared data library.
In the top menu bar, go to
- Click on
Data Libraries . - Click on
Galaxy Australia Training Material: Sample Identification . - Tick the box next to the four files.
- Click the
To History button, select As Datasets. - Name a new history and click
Import . - In the top menu bar, click
Analyze Data . - You should now have four files in your current history.
Or, import from the web
Only follow this step if unable to load the data files from shared data, as described above.
- In a new browser tab, go to this webpage:
![]()
- Right click on a name: select “copy link address”
- In Galaxy, go to
Get Data and then Upload File - Click
Paste/Fetch data - A box will appear: paste in link address
- Click
Start - Click
Close - The file will now appear in the top of your history panel.
- Repeat this for all the files on Zenodo.
Shorten file names
- Click on the pencil icon next to the file name.
- In the centre Galaxy panel, click in the box under
Name - Shorten the file name.
- Then click
Save
You should now have four files in your current history:
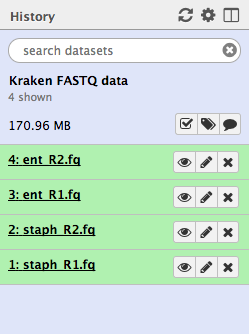
Run Kraken
We have a sample that should be Staphylococcus aureus. The paired-end FASTQ read files are:
staph_R1.fq andstaph_R2.fq .
(We will look at the other set of files later on in the tutorial).
-
Go to
Tools search bar and type in “Kraken”. Click on the Kraken tool. -
Set the following parameters:
Single or paired reads : PairedForward strand: staph_R1.fq Reverse strand: staph_R2.fq Select a Kraken database: Minikraken- leave other settings as they are
- Click
Execute
Examine the output
The output is a file called
Click
When the file is green, click on the eye icon to view.
- We will turn this output into something easier to read in the next step.
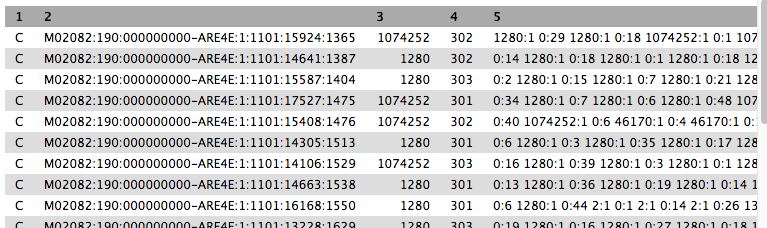
- Column 2 is the sequence ID.
- Column 3 is the taxon ID (from NCBI).
- Column 5 is a summary of all the taxon IDs that each k-mer in the sequence matched to (taxon ID:number of k-mers).
Kraken report
-
Go to
Tools search bar and type in “Kraken-report”. Click on the Kraken-report tool. -
Set the following parameters:
Kraken output :Kraken on data x and x: Classification Select a Kraken database : Minikraken- Click
Execute
The output file is called
- Click on the eye icon to view.
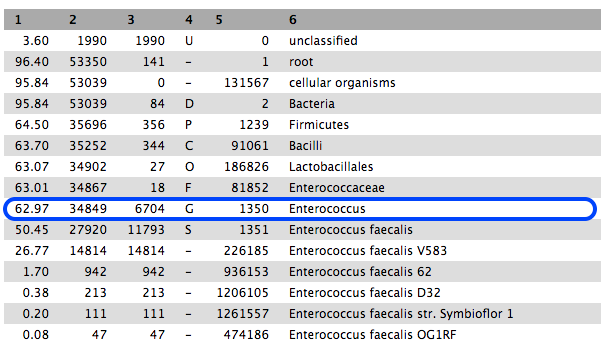
- Column 1: percentage of reads in the clade/taxon in Column 6
- Column 2: number of reads in the clade.
- Column 3: number of reads in the clade but not further classified.
- Column 4: code indicating the rank of the classification: (U)nclassified, (D)omain, (K)ingdom, (P)hylum, (C)lass, (O)rder, (F)amily, (G)enus, (S)pecies).
- Column 5: NCBI taxonomy ID.
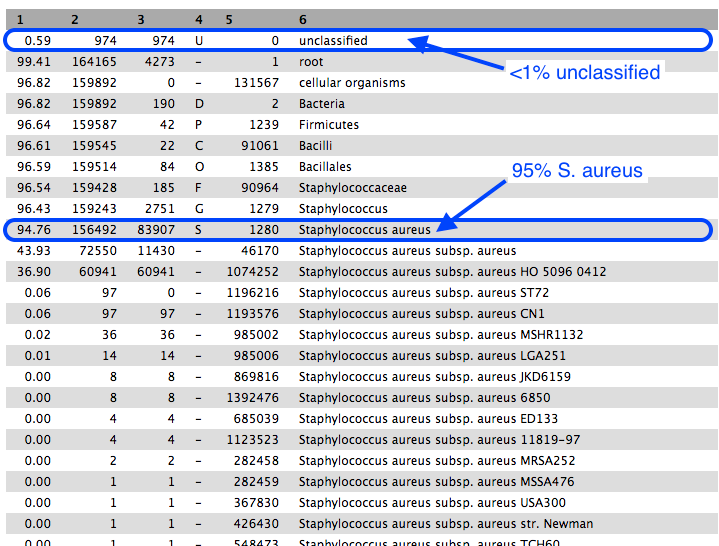
Approximately 95% of reads were classified as Staphylococcus aureus, confirming the correct identity of our bacterial sample.
- Of these reads, roughly half were uniquely present in S. aureus subsp. aureus, and most of those were uniquely present in strain HO 5096 0412.
- The sample strain is therefore most related to the HO 5096 0412 strain.
The remaining reads within the S. aureus clade were classified into various taxa.
- Scroll down column 3 to see the number of reads assigned directly to the taxon in column 6.
- These are all very low and can be disregarded.
Next
Re-run Kraken with another sample. This sample should be Enterococcus faecalis.
- Use the files
ent_R1.fq andent_R2.fq . - Run
Kraken with these files. These are paired-end reads. - With the
Classification file from Kraken, runKraken-report . - Cick on the eye icon to view the
Kraken-report file.
-
63% are classified to the genus Enterococcus, and most of these to E. faecalis.
-
However, if we scroll down the table of results, we see that 31% are classified to the genus Mycobacterium, mostly M. abscessus. These are not in the same phylum as Enterococcus.
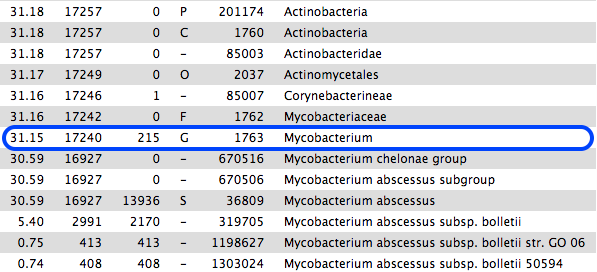
- This sample is probably contaminated.
Links
See this history in Galaxy
If you want to see this Galaxy history without performing the steps above:
- Log in to Galaxy Australia: https://usegalaxy.org.au/
- Go to
Shared Data - Click
Histories - Click
Completed-kraken-analysis - Click
Import (at the top right corner) - The analysis should now be showing as your current history.
What’s next?
To use the tutorials on this website:
- ← see the list in the left hand panel
- ↖ or, click the menu button (three horizontal bars) in the top left of the page
You can find more tutorials at the Galaxy Training Network: