Workflow: RNA-Seq
Create a Galaxy workflow
Galaxy provides the option to extract a workflow based on the steps you just followed and the tools and parameters used.
RNA-Seq
- Log in to your Galaxy instance (for example, Galaxy Australia, usegalaxy.org.au).
Before we create a workflow for RNA-seq, we need the correct files in our current Galaxy history. We can get these files in two ways:
-
Either follow the tutorial on RNA-seq
-
Or, obtain the files from a Shared History (Galaxy Australia only).
- Go to
Shared Data - Click
Histories - Click
Published-RNA-seq-bacteria - Click
Import (at the top right corner) - The analysis should now be showing as your current history.
- Go to
When you have completed this tutorial or imported the history, you will have the correct files in your history panel.
Extract the Workflow
- In the history panel, click on the cog icon.
- Click
Extract Workflow - If all looks ok, click
Create Workflow - In the centre panel, click
edit
This brings up the workflow canvas, a space for graphically viewing and re-arranging your workflow.
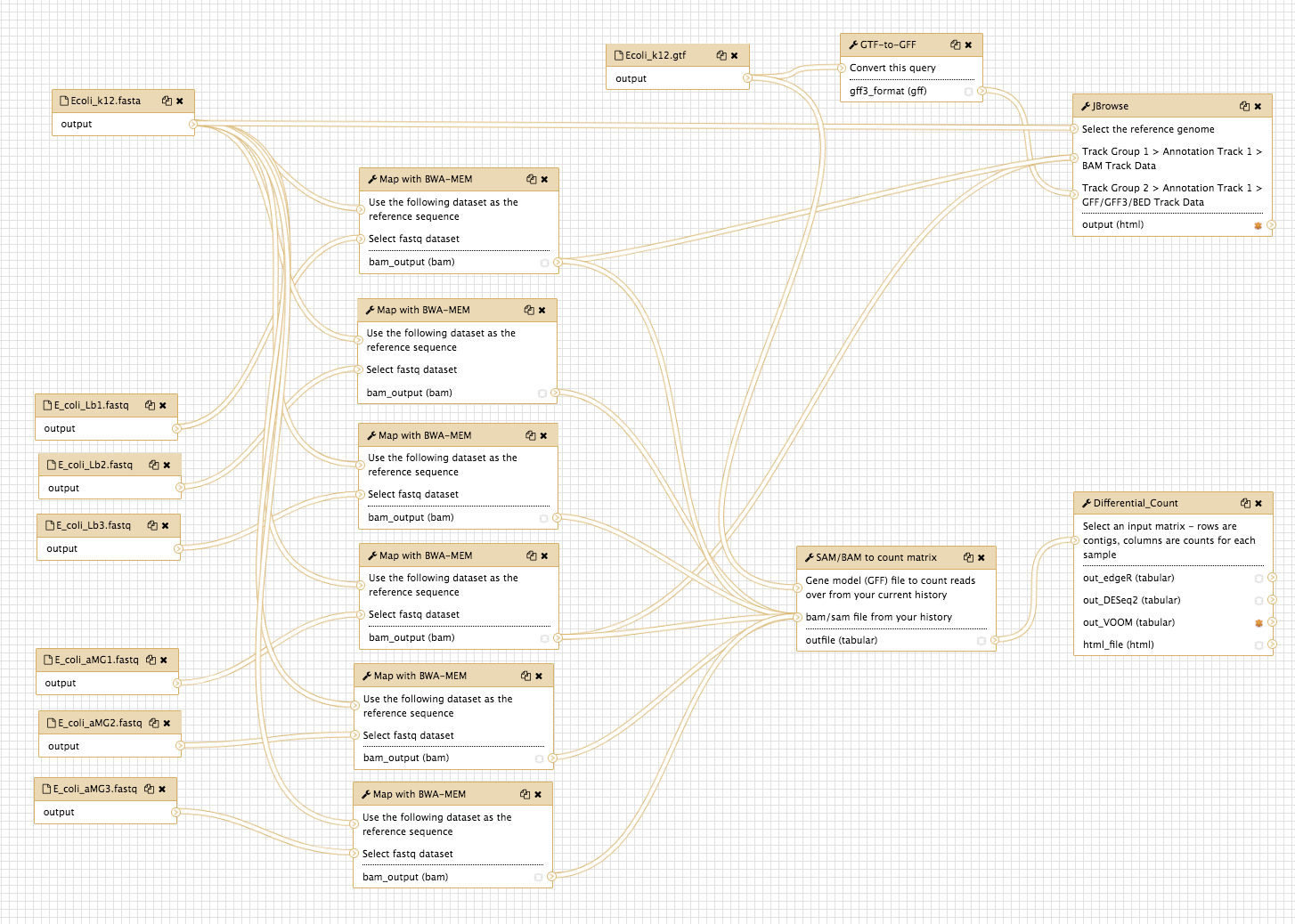
- Drag the panes around until your workflow is clear.
- Click on the star icon next to any output files that you want to retain, such as the JBrowse output and the voom output.
- In the top right corner click on the cog icon and
Save -
Click on the cog icon again and
Run Send results to a new history : Yes- Check all input files are correct and change if necessary
- In the top right corner
Run workflow
-
This workflow will now run.
- In the History panel, click on the
View all histories icon
- Find your workflow history and
Switch to it.
To see all your workflows, go to the top panel in Galaxy and click
- You can edit or run workflows from here by clicking on the drop-down arrow next to each workflow.
What’s next?
To use the tutorials on this website:
- ← see the list in the left hand panel
- ↖ or, click the menu button (three horizontal bars) in the top left of the page
You can find more tutorials at the Galaxy Training Network: