Variant calling with Snippy
Variant calling is the process of identifying differences between two genome samples.
- Differences are usually limited to single nucleotide polymorphisms (SNPs) and small insertions and deletions (indels).
- Larger structural variation such as inversions, duplications and large deletions are not typically covered by “variant calling”.
- In this tutorial, we will use the tool “Snippy” (link to Snippy is here).
- Snippy uses a tool to align the reads to a reference genome, and another tool to decide (“call”) if the discrepancies are real variants.
Get data
- Log in to your Galaxy instance (for example, Galaxy Australia, usegalaxy.org.au).
Use shared data
If you are using Galaxy Australia, you can import the data from a shared data library.
In the top menu bar, go to
- Click on
Data Libraries . - Click on
Galaxy Australia Training Material: Variant Calling: Microbial Variant Calling . - Tick the boxes next to the five files.
- Click the
To History button, select As Datasets. - Name a new history and click
Import . - In the top menu bar, click
Analyze Data . - You should now have five files in your current history.
Or, import from the web
Only follow this step if unable to load the data files from shared data, as described above.
- In a new browser tab, go to this webpage:
![]()
- Find the file called
mutant_R1.fastq - Right click on file name: select “copy link address”
- In Galaxy, go to
Get Data and then Upload File - Click
Paste/Fetch data - A box will appear: paste in link address
- Click
Start - Click
Close - The file will now appear in the top of your history panel.
- Shorten the file name if you wish. (Click the pencil icon).
Repeat these steps for the other set of reads (reverse reads) called
- In a new browser tab, go to this webpage:
![]()
- Find the file called
mutant_R2.fastq - Right click on file name: select “copy link address”
- In Galaxy, go to
Get Data and then Upload File - Click
Paste/Fetch data - A box will appear: paste in link address
- Click
Start - Click
Close - The file will now appear in the top of your history panel.
- Shorten the file name if you wish.
Repeat these steps for three other files:
Shorten file names
- Click on the pencil icon next to the file name.
- In the centre Galaxy panel, click in the box under
Name - Shorten the file name.
- Then click
Save
Quality Control
If you want to check the quality of your reads, see the Quality Control tutorial.
- Note: Skip over the “Import Data” section and instead use the file called
mutant_R1.fastq that is already in your current history.
Call variants with Snippy
Go to the Tool panel and search for “snippy” in the search box.
- Click on
snippy
Set the following parameters (leave other settings as they are):
- For
Reference type select Genbank. - Then for
Reference Genbank choose thewildtype.gbk file. - For
Single or Paired-end reads choose Paired. - Then choose the first set of reads,
mutant_R1.fastq and second set of reads,mutant_R2.fastq . - Click
Advanced Parameters and forOutput selection , select all the files.
Your tool interface should look a bit like this (not exactly, as tool versions are often updated):
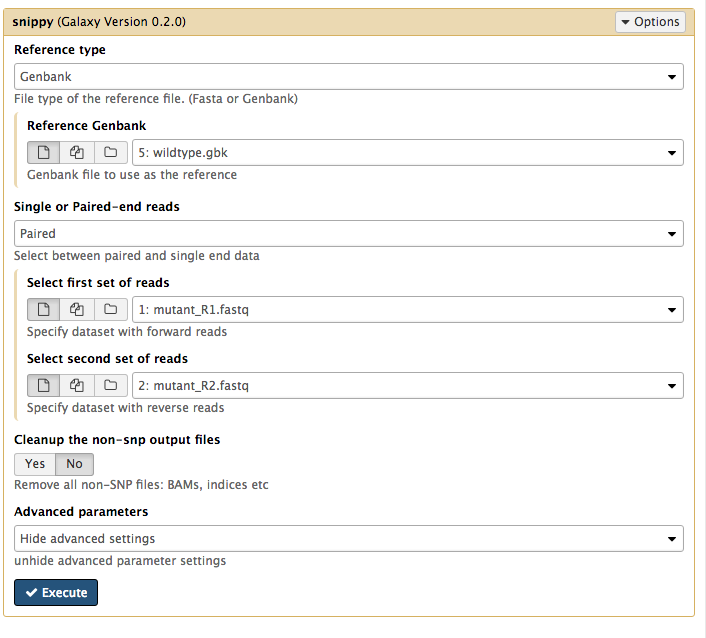
- Click
Execute .
Examine Snippy output
From Snippy, there are 10 output files in various formats.
- Go to the table file called
snippy on data XX, data XX and data XX table and click on the eye icon. - (The XX refers to the number that Galaxy has given your files.)
- We can see a list of variants. Look in column 3 to see which types the variants are, such as a SNP or a deletion.
- Look at the third variant called. This is a T→A mutation, causing a stop codon. Look at column 14: the product of this gene is a methicillin resistance protein. Methicillin is an antibiotic. What might be the result of such a mutation?
View Snippy output in JBrowse
-
Go to the Galaxy tools panel, and use the search box at the top to search for “JBrowse”.
-
Under
Reference genome to display choose Use a genome from history. -
Under
Select the reference genome choosewildtype.fna . This sequence will be the reference against which annotations are displayed. -
For
Produce a Standalone Instance select Yes. -
For
Genetic Code choose 11: The Bacterial, Archaeal and Plant Plastid Code. -
Under
JBrowse-in-Galaxy Action choose New JBrowse Instance. -
We will now set up three different tracks - these are datasets displayed underneath the reference sequence (which is displayed as nucleotides in FASTA format). We will choose to display the sequence reads (the .bam file), the variants found by snippy (the .gff file) and the annotated reference genome (the wildtype.gff)
Track 1 - sequence reads
- Click
Insert Track Group - For
Track Cateogry name it “sequence reads” - Click
Insert Annotation Track - For
Track Type choose BAM Pileups - For
BAM Track Data selectthe snippy bam file - For
Autogenerate SNP Track select Yes
Track 2 - variants
- Click
Insert Track Group again - For
Track Category name it “variants” - Click
Insert Annotation Track - For
Track Type choose GFF/GFF3/BED/GBK Features - For
Track Data selectthe snippy snps gff file
Track 3 - annotated reference
- Click
Insert Track Group again - For
Track Category name it “annotated reference” - Click
Insert Annotation Track - For
Track Type choose GFF/GFF3/BED/GBK Features - For
Track Data selectwildtype.gff
- Click
Execute
A new file will be created, called
-
Click on the eye icon next to the file name. The JBrowse window will appear in the centre Galaxy panel.
-
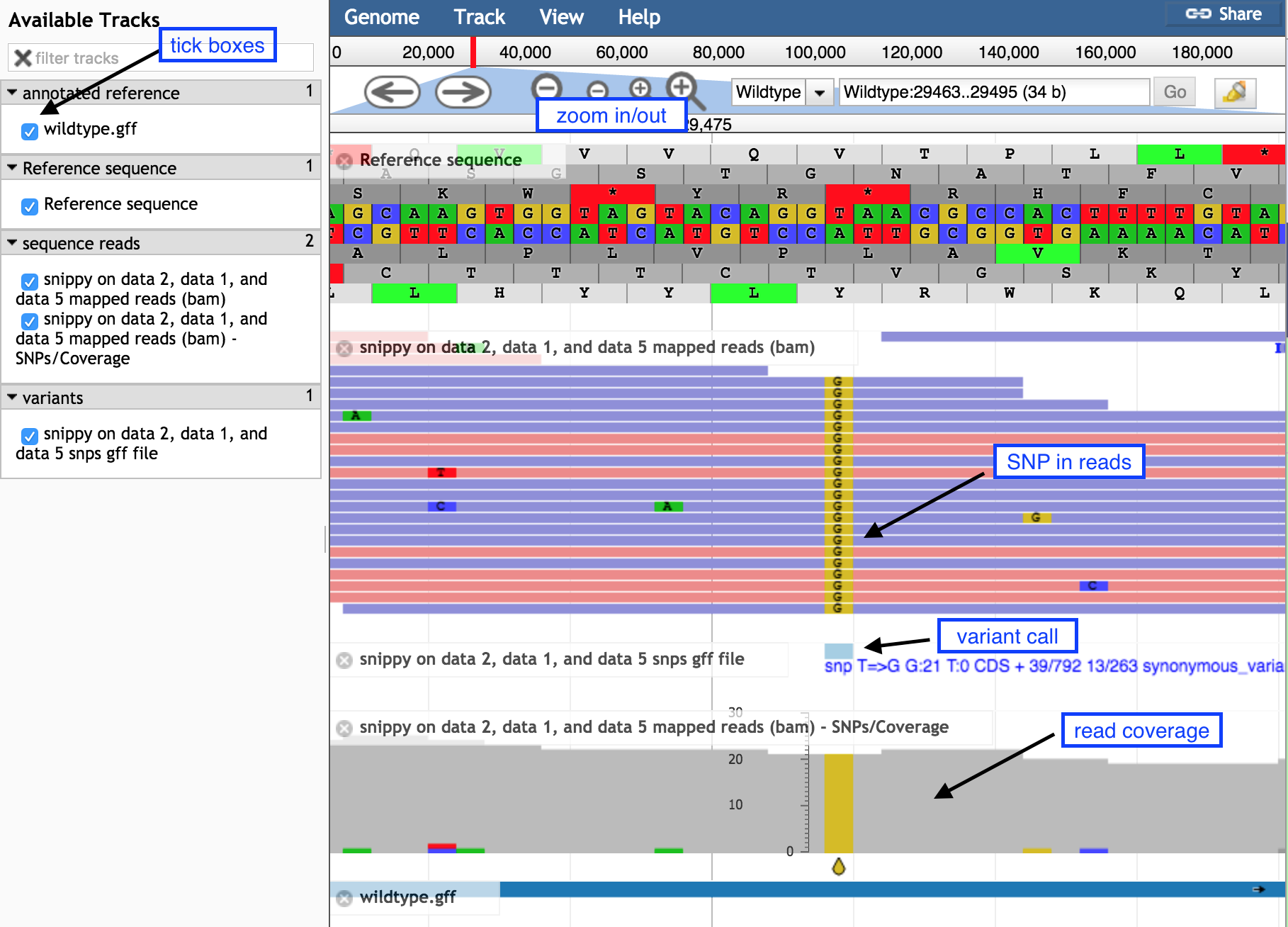
On the left, tick boxes to display the tracks
-
Use the minus button to zoom out to see:
- sequence reads and their coverage (the grey graph)
-
Use the plus button to zoom in to see:
- probable real variants (a whole column of snps)
- probable errors (single one here and there)
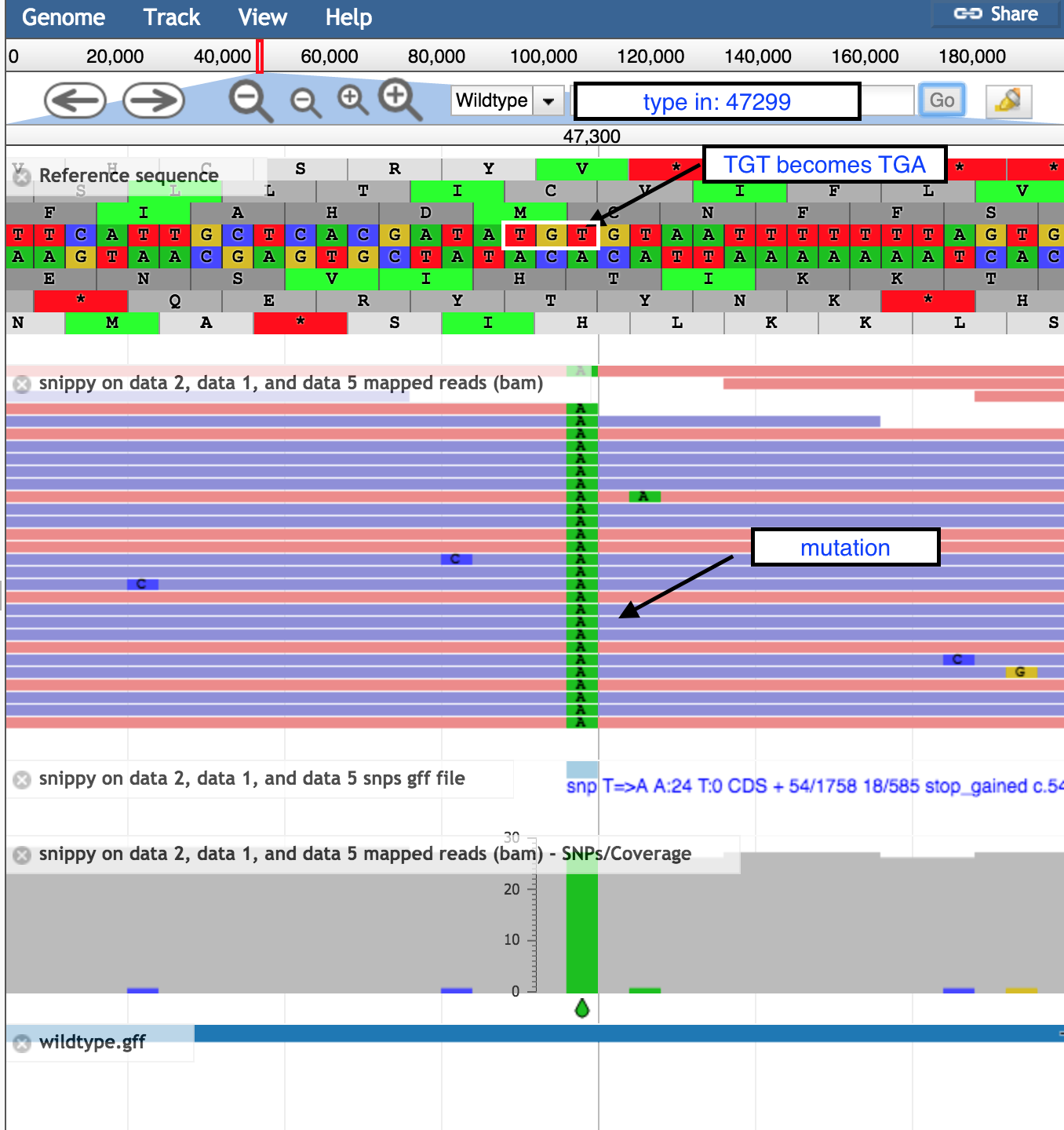
- In the coordinates box, type in 47299 and then
Go to see the position of the SNP discussed above.- the correct codon at this position is TGT, coding for the amino acid Cysteine, in the middle row of the amino acid translations.
- the mutation of T → A turns this triplet into TGA, a stop codon.
Extension exercise
Let’s repeat this tutorial using other data.
- We will investigate the bacteria Mycoplasma synoviae as it has a small genome and will not take too long to analyse.
- We want to find out if our sequencing reads vary from a reference genome.
-
To get some sequencing reads, follow the instructions here.
- This will give us paired-end sequencing reads (two files).
-
These files are very large. To subsample them so that we can run the analysis in a shorter time:
- Search for Select first in the tool search box
- Select first 4001 lines
- from the
R1.fastq file - Execute
- Repeat this for the
R2.fastq file
-
To obtain a reference genome, follow the instructions here including the Prokka step.
- This will give us 12 output files from Prokka.
- We need three of these files: the genbank file
Prokka.gbk , the fasta fileProkka.fna , and the gff fileProkka.gff . - Galaxy has interpreted the genbank file as a text file. Click on the pencil icon next to the genbank file; click Datatypes, and change the datatype to “genbank”.
-
Run snippy on the sequencing reads.
- For the reference genome, use the
Prokka:gbk file. - Remember to include all the output files.
- For the reference genome, use the
- Run JBrowse to produce a visualization of the called variants.
- For the reference genome, use the
Prokka.fna file. - As we did above, set up three “Tracks” to view underneath this reference genome nucleotide sequence: The
snippy.bam file showing the reads mapped to the reference, thesnippy.gff file showing the snps (the variants), and the annotated reference genomeProkka.gff .
- For the reference genome, use the
You should now have a JBrowse file showing the results of variant calling on these reads of the bacteria Mycoplasma synoviae. Can you find any variants?
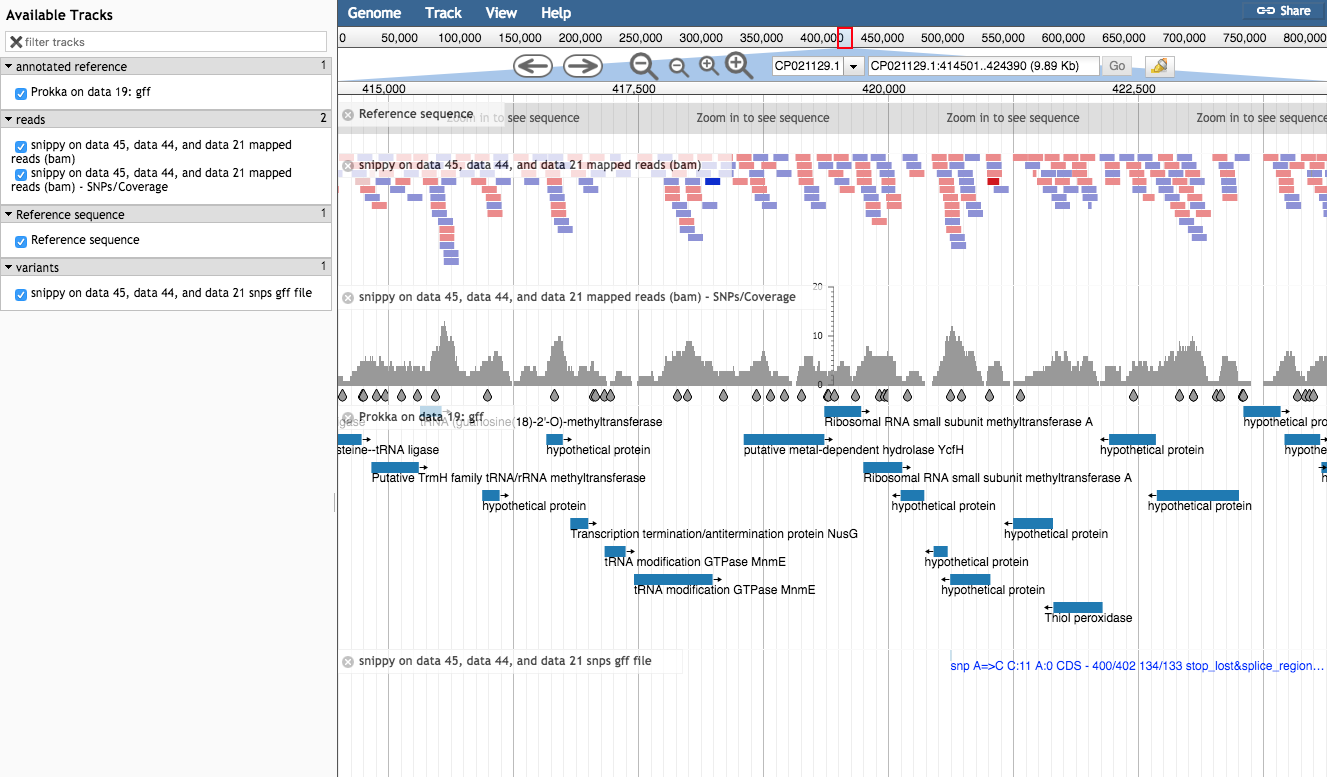
The workflow:
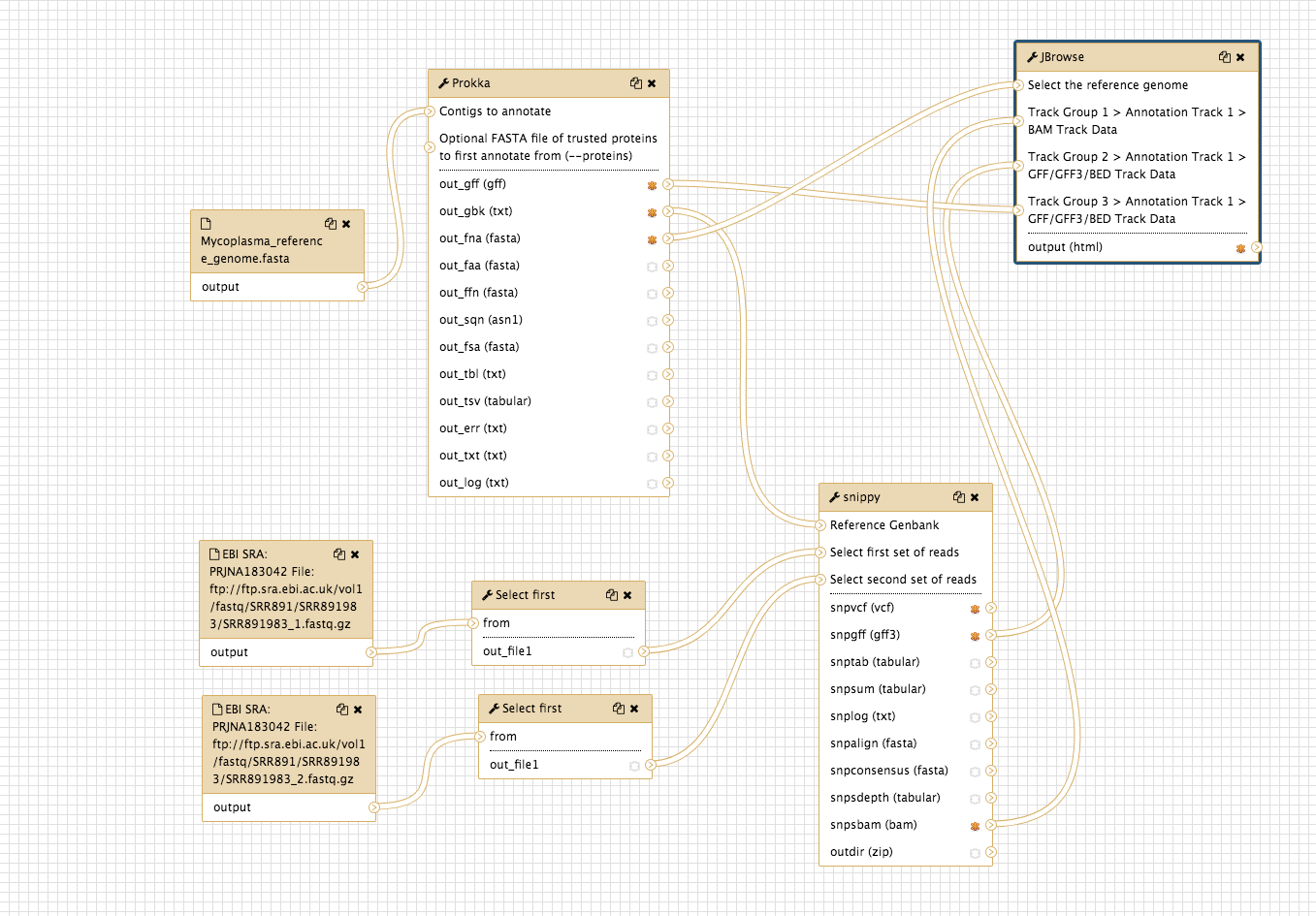
See this history in Galaxy
If you want to see this Galaxy history without performing the steps above:
- Log in to Galaxy Australia: https://usegalaxy.org.au/
- Go to
Shared Data - Click
Histories - Click
Variant_calling_with_snippy - (or, for the extension part, click
Variant-calling-bacteria-extension ) - Click
Import (at the top right corner) - The analysis should now be showing as your current history.
More information
Here are some references covering more information about variant calling.
** Comparison of variant calling tools: ** Sandmann S, de Graaf AO, Karimi M, van der Reijden BA, Hellström-Lindberg E, Jansen JH, Dugas M. Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Sci Rep. 2017 Feb 24;7:43169.
** Human variant calling pipelines: ** Hwang S, Kim E, Lee I, Marcotte EM. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep. 2015 Dec 7;5:17875.
** Rare and common disease variants: ** Fritsche LG, Igl W, Bailey JNC, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016 Feb;48(2):134–43.
** Variant calling in the Ebola virus: ** Quick J, Loman NJ, Duraffour S, Simpson JT, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016 Feb 11;530(7589):228–32.
** Variant calling to detect antibiotic-resistant bacteria: ** Kpeli G, Buultjens AH, Giulieri S, Owusu-Mireku E, Aboagye SY, Baines SL, Seemann T, Bulach D, Gonçalves da Silva A, Monk IR, Howden BP, Pluschke G, Yeboah-Manu D, Stinear T. Genomic analysis of ST88 community-acquired methicillin resistant Staphylococcus aureus in Ghana. PeerJ. 2017 Feb 28;5:e3047.
** High coverage artifacts: ** Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014 Oct 15;30(20):2843–51.
What’s next?
To use the tutorials on this website:
- ← see the list in the left hand panel
- ↖ or, click the menu button (three horizontal bars) in the top left of the page
You can find more tutorials at the Galaxy Training Network: